Edit distance is a fundamental metric in computer science and bioinformatics that quantifies how dissimilar two strings are by counting the minimum number of operations required to transform one string into another. The most common operations include:
For example, the edit distance between "kitten" and "sitting" is 3:
kitten → sitten (substitute 'k' with 's')sitten → sittin (substitute 'e' with 'i')sittin → sitting (insert 'g')Traditional dynamic programming algorithms for computing edit distance, such as the Wagner-Fischer algorithm, have a time and space complexity of O(mn), where m and n are the lengths of the two strings. This becomes impractical for long sequences, such as DNA strands in bioinformatics.
The Wavefront Alignment (WFA) algorithm optimizes space usage by focusing on the "wavefront" of the dynamic programming table, only computing and storing necessary parts. It exploits the fact that edits occur in diagonal patterns across the table, allowing for significant reductions in both time and space complexity.
Importantly, the WFA algorithm uses O(ns) space, where n is the sequence length and s is the maximum edit distance. This parameterized linear space requirement makes it suitable for aligning long sequences efficiently.
// Initialize wavefront arrays
M_old = [INT_MIN] * (2 * max_score + 1)
M_new = [INT_MIN] * (2 * max_score + 1)
M_old[offset] = 0
score = 0
while true:
for k from -score to score:
// Calculate insertion, deletion, and substitution
ins = M_old[offset + k - 1] + 1 if k > -score else INT_MIN
del = M_old[offset + k + 1] + 1 if k < score else INT_MIN
sub = M_old[offset + k]
max_val = max(ins, del, sub)
if max_val == INT_MIN:
continue
// Diagonal extension
i_pos = max_val
j_pos = i_pos - k
while i_pos < m and j_pos < n and str1[i_pos] == str2[j_pos]:
i_pos += 1
j_pos += 1
M_new[offset + k] = i_pos
// Check for alignment completion
if i_pos >= m and j_pos >= n:
return score
// Swap wavefronts for next iteration
M_old, M_new = M_new, [INT_MIN] * (2 * max_score + 1)
score += 1
The following is the C++ implementation of the Wavefront Alignment algorithm without vectorization:
int wavefront_alignment_sequential(const string& str1, const string& str2) {
int m = str1.length();
int n = str2.length();
int max_score = m + n;
vector<int> M_old(2 * max_score + 1, INT_MIN);
vector<int> M_new(2 * max_score + 1, INT_MIN);
int offset = max_score;
M_old[offset] = 0;
int score = 0;
while (true) {
for (int k = -score; k <= score; ++k) {
int diag_idx = offset + k;
if (diag_idx < 0 || diag_idx >= 2 * max_score + 1) {
continue;
}
int ins = (k > -score) ? M_old[diag_idx - 1] + 1 : INT_MIN;
int del = (k < score) ? M_old[diag_idx + 1] + 1 : INT_MIN;
int sub = M_old[diag_idx];
int max_val = max({ins, del, sub});
if (max_val == INT_MIN) continue;
int i_pos = max_val;
int j_pos = i_pos - k;
while (i_pos < m && j_pos < n && str1[i_pos] == str2[j_pos]) {
++i_pos;
++j_pos;
}
M_new[diag_idx] = i_pos;
if (i_pos >= m && j_pos >= n) {
return score;
}
}
M_old.swap(M_new);
fill(M_new.begin(), M_new.end(), INT_MIN);
++score;
}
}
Below is the AVX-512 optimized version of the WFA algorithm. It leverages SIMD instructions to process multiple diagonals in parallel:
int wavefront_alignment_avx512(const string& str1, const string& str2) {
int m = str1.length();
int n = str2.length();
int max_score = m + n;
vector<int> M(2 * max_score + 1, INT_MIN);
int offset = max_score;
M[offset] = 0;
int score = 0;
while (true) {
for (int k = -score; k <= score; k += 16) {
int remaining_diagonals = min(16, score - k + 1);
__mmask16 valid_mask = (1 << remaining_diagonals) - 1;
__m512i diag_indices = _mm512_set_epi32(
k + 15, k + 14, k + 13, k + 12, k + 11, k + 10, k + 9, k + 8,
k + 7, k + 6, k + 5, k + 4, k + 3, k + 2, k + 1, k);
__m512i M_curr = _mm512_maskz_loadu_epi32(valid_mask, &M[offset + k]);
__m512i M_left = _mm512_maskz_loadu_epi32(valid_mask, &M[offset + k - 1]);
__m512i ins = _mm512_mask_add_epi32(M_curr, valid_mask, M_left, _mm512_set1_epi32(1));
__m512i M_right = _mm512_maskz_loadu_epi32(valid_mask, &M[offset + k + 1]);
__m512i del = _mm512_mask_add_epi32(M_curr, valid_mask, M_right, _mm512_set1_epi32(1));
__m512i sub = M_curr;
__m512i max_val = _mm512_max_epi32(ins, _mm512_max_epi32(del, sub));
alignas(64) int max_values[16];
_mm512_store_epi32(max_values, max_val);
for (int i = 0; i < remaining_diagonals; ++i) {
int diag = k + i;
int diag_idx = offset + diag;
int i_pos = max_values[i];
int j_pos = i_pos - diag;
while (i_pos < m && j_pos < n && str1[i_pos] == str2[j_pos]) {
++i_pos;
++j_pos;
}
max_values[i] = i_pos;
M[diag_idx] = i_pos;
if (i_pos >= m && j_pos >= n) {
return score;
}
}
_mm512_mask_storeu_epi32(&M[offset + k], valid_mask, _mm512_load_epi32(max_values));
}
++score;
}
}
We conducted experiments using synthetic DNA sequences generated with a controlled mutation rate:
The results of the experiments are as follows:
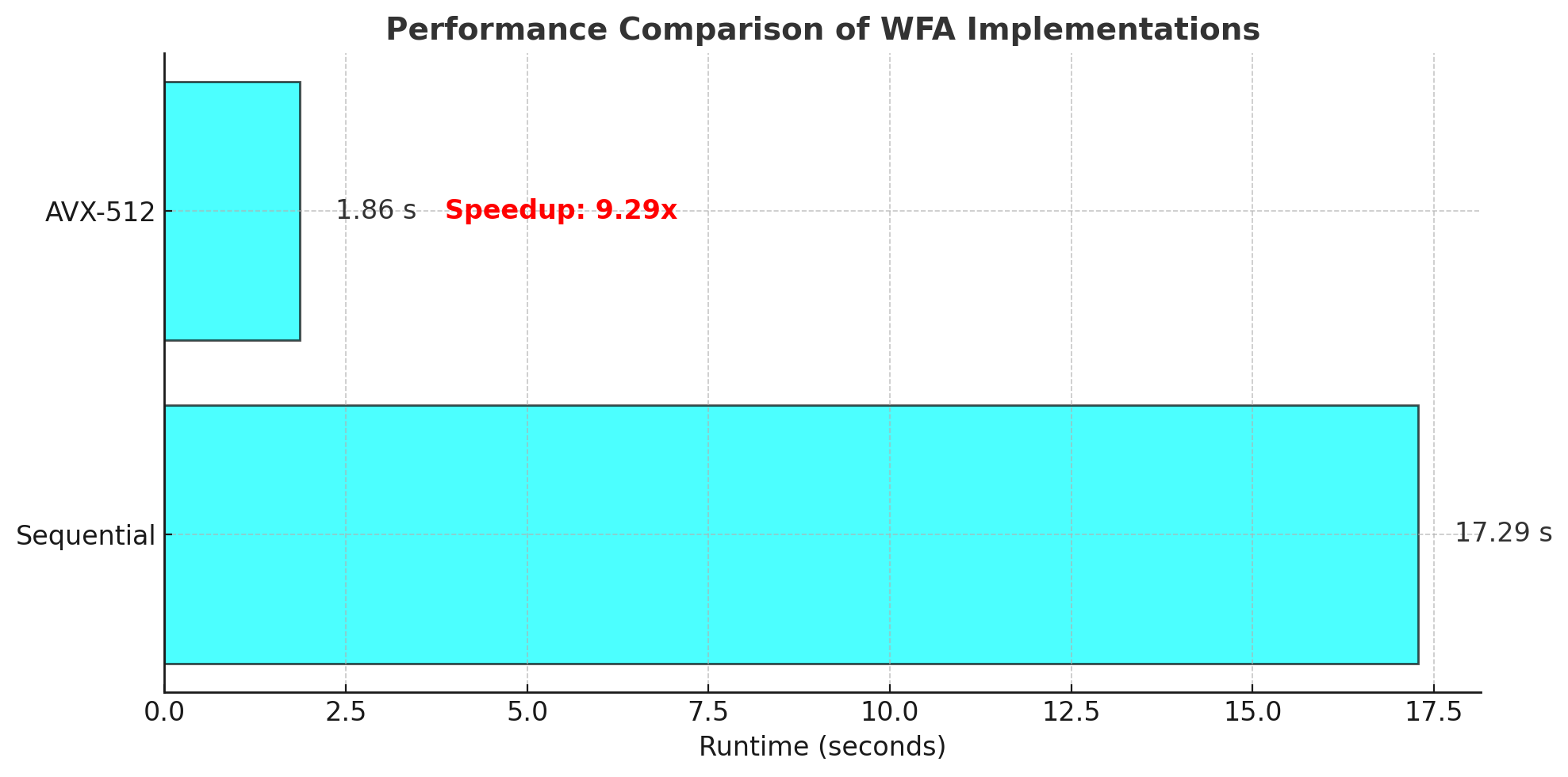
The above plot illustrates the performance of the sequential and AVX-512 implementations. The horizontal bars represent the runtime in seconds, and the transparent cyan color highlights the difference in execution time.
The AVX-512 implementation achieves a significant speedup of 9.29x over the sequential version. This is attributed to the following factors:
-O3 compiler flag optimizes both implementations, but the AVX-512 version benefits more due to explicit vectorization.The Wavefront Alignment algorithm provides an efficient way to compute edit distances with reduced space complexity, using O(ns) space where n is the sequence length and s is the maximum edit distance. Leveraging AVX-512 instructions further accelerates the computation by parallelizing operations across multiple data points. Our experiments demonstrate substantial performance gains on modern CPUs, with a speedup of 9.29x over the sequential implementation.
While AVX-512 offers significant acceleration, the parameterized linear space requirements of the WFA algorithm make it well-suited for GPUs, which can handle large-scale parallel computations more efficiently. As data sizes continue to grow, combining algorithmic efficiency with hardware capabilities becomes increasingly important.
Marco-Sola, Santiago, Moure, Juan Carlos, Moreto, Miquel, & Espinosa, Antonio. Fast gap-affine pairwise alignment using the wavefront algorithm. Bioinformatics, 37(4), 456–463. Oxford University Press, 2020. http://dx.doi.org/10.1093/bioinformatics/btaa777.